La rétinite pigmentaire
Lorsque la rétine périphérique est pigmentée et qu’il y a un rétrécissement progressif du champ visuel, lorsque l’on constate une gêne de la vision nocturne, on peut se trouver en présence d’une rétinite pigmentaire. La rétinite pigmentaire est une maladie qui touche en France environ 40 000 personnes, un chiffre pouvant être doublé si l’on y associe les autres maladies génétiques. Contrairement à la dégénérescence maculaire, c’est la rétine périphérique qui va être détruite dans l’affection.
Signes cliniques
L’héméralopie est le premier signe ressenti par le patient. Il s’agit d’une mauvaise vision dès que la lumière est un peu faible. Elle apparaît chez le sujet très jeune. Le deuxième élément étant la baisse progressive du champ visuel qui va rétrécir. Les symptômes sont bilatéraux. L’évolution va être très variable dans le temps et dans la gravité, chaque malade ayant une évolution particulière.

vue d’une rétinite évoluée
Quoique certains cas restent isolés, le caractère familial de l’affection est souvent retrouvé. La transmission peut être autosomique dominante, autosomique récessive ou encore liée à l’X. Dans ce dernier cas, la maladie est plus grave et de début plus précoce.
Les examens complémentaires
L’acuité visuelle est excellente pendant très longtemps, l’affection ne touchant pas la région maculaire centrale. Le champ visuel est très rapidement rétréci. L’examen de la rétine, après dilatation de la pupille, va montrer des petits dépôts noirâtres en périphérie rétinienne. Il s’y associe un rétrécissement du calibre des artères et apparition rapide d’une pâleur papillaire. Un élément de diagnostic est primordial. Il s’agit de « l’électrorétigramme » correspondant à l’activité de la rétine.
Cet électrorétigramme est très précocement altéré avant même parfois que n’apparaissent les premiers signes de l’affection. La personne touchée devient extrêmement gênée : en fin évolution, son champ visuel est rétréci à un mince filet central puis c’est à son tour la macula qui va être altérée avec la chute de l’acuité visuelle centrale qui marque le point final de l’évolution.

Vue d’une rétinite ultime
Formes cliniques
Il existe de nombreuses variantes, soit dans l’aspect anatomique, soit dans l’aspect clinique. Les formes unilatérales sont très rares. Il existe des formes où l’on observe, à l’examen de fond de l’œil, des petites tâches blanchâtres. il s’agit alors de rétinite ponctuée albescente. On peut rapprocher également « l’amaurose congénitale de Leber » qui entraîne une quasi cécité dès la première année de vie.
Ailleurs, la rétinophatie pigmentaire est associée à d’autres pathologies :
– Dans le syndrome de Uscher, une surdité est associée à la rétinite pigmentaire. Ce syndrome se voit dans 15 % de l’ensemble des rétinopathies pigmentaires.
– Le syndrome de Bardet-Bield associe une rétinite pigmentaire, un retard mental, une obésité, un hypogonadisme et une déformation des doigts. L’affection se transmet sur le mode autosomique récessif.
– Dans le Fundus Albipuncatus, on observe, à l’examen du fond de l’œil, de nombreux points jaunâtres, la cécité nocturne congénitale est précoce.
Origine de l’affection
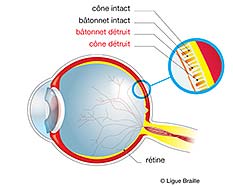
Dans la rétinite pigmentaire, ce sont les bâtonnets, cellules visuelles occupant la majeure partie de la rétine mais absentes de la zone centrale maculaire, qui sont déficientes. Elles vont peu à peu dégénérer entraînant une baisse du champ visuel. Il n’existe aucune thérapeutique pour traiter ces maladies.
Un traitement par vitamine A peut retarder l’évolution de l’affection, de même le port de verres teintés éliminant la lumière bleue et verte. Pour l’avenir, les espoirs se tournent vers la « thérapie génique ». Dans la rétinopathie de Leber, des chiens brillards, une race qui présente la même affection, ont pu être guéris, avec arrêt de l’évolution de la maladie, lorsqu’on injectait sous la rétine le gène manquant lié à un adénovirus.
Ce traitement représente une avancée considérable. L’étude chez l’homme pourra être envisagée lorsqu’on sera certain de l’innocuité des virus utilisés et lorsqu’on connaîtra parfaitement les modalité d’action de la thérapie génique.
Actuellement, nous n’avons malheureusement aucune possibilité pour traiter les maladies rétiniennes génétiques du type rétinopathie pigmentaire. Cependant ces maladies doivent être diagnostiquées aussi rapidement que possible. Elles doivent faire l’objet de recherches très coûteuses en moyens humains et matériels. Seules ces recherches permettront de les guérir dans quelques années.
(Professeur Bernard Arnaud Chef de service d’ophtalmologie, Centre Hospitalier Gui de Chauliac, Montpellier)